How to Submit
Submitting Your Research for Review
As of December 5, 2025 the HSRO is no longer accepting emailed submissions - we have moved to the IRB Module in Novelution. Please login to Novelution to submit your research.
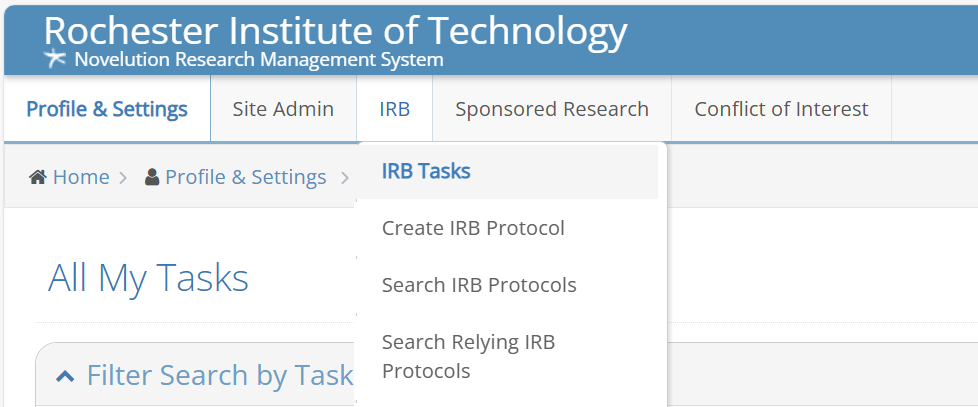
Once you are logged-in you will see a menu across the top; go to IRB, click on Create IRB Protocol and you can begin the process of developing your submission.
Training opportunities will be announced here on the website as well as through your Department via email.
HSRO Review
Submissions go through an initial review in the HSRO. Your submission is reviewed for completeness and clarity. If there are any questions or missing items the Investigator will be contacted via comments in the Novelution record for additional information.
- If the necessary criteria for approval are met, research that is considered exempt will be approved after the initial review, and an approval letter emailed to the investigator and data collection can begin. You can follow your protocol's progress in Novelution by viewing the Requirements panel of your submission.
- Research that is considered expedited may be reviewed by experienced reviewers as well as the HSRO. This is set up at the convenience of the reviewer’s schedule and will take additional time after the initial review by the HSRO. After review, the research will either be approved, deferred pending additional information, or deferred to the entire Board at a convened monthly meeting.
- Research that is deferred pending additional information will be approved once all of the questions and concerns have been addressed and an approval letter sent to the Investigator.
- Research that is deferred to a Board meeting will be discussed at a convened Board meeting; after review it will either be approved, deferred pending additional information, or disapproved. If it is approved an approval letter is sent to the investigator and data collection can begin. If it is deferred, the investigator will need to submit additional information and then it will be reviewed again. If it is disapproved, the investigator can submit with revisions and may request a conversation with the HSRO or the Board.
Amendments
Approved research projects may need to be modified; this can be accomplished by requesting an amendment in the Novelution record. Any modifications must be reviewed and approved by the HSRO before being implemented. Examples of modifications include changes in study staff, study procedures, consent processes, recruitment materials and study sites.
Steps to Amending Research
Go into the protocol and click on the Request button in the New Amendment panel.
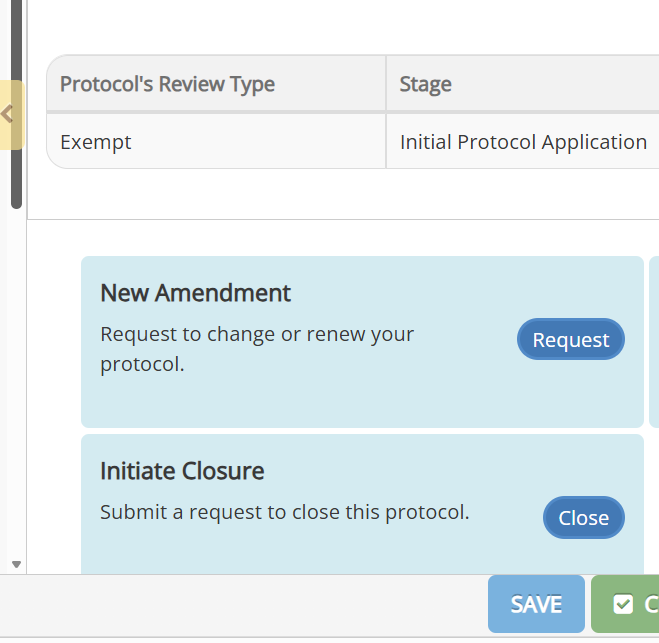
Update to reflect the proposed changes. For projects submitted and approved by the HSRO prior to the implementation of Novelution you will need to first submit your project for in Novelution to create a record that you can go in and amend.
Continuing Review
An IRB will conduct continuing review of research at intervals appropriate to the degree of risk, not less than once per year. Unless an IRB determines otherwise, continuing review of research is not required in the following circumstances:
- Research eligible for expedited review in accordance with §46.110;
- Research reviewed by the IRB in accordance with the limited IRB review described in §46.104(d)(2)(iii);
- Research that has progressed to the point that it involves only one or both of the following, which are part of the IRB-approved study:
- Data analysis, including analysis of identifiable private information or identifiable biospecimens, or
- Accessing follow-up clinical data from procedures that subjects would undergo as part of clinical care.
If continuing review is required, the your approval letter will indicate the date continuing review must take place by. Investigators are responsible for submitting within two months of the one year anniversary date. It is the responsibility of the investigator to ensure that continuing review is up to date.
If continuing review is indicated, IRB approval expires automatically at the one year anniversary date.
What the IRB Expects in a Submission
The Institutional Review Board is an administrative body established to safeguard the rights and welfare of all human subjects who participate in research projects conducted by RIT. In compliance with federal law and institutional policy, all research projects involving human subjects or human material must be reviewed and approved by the IRB.
The overall criteria for IRB approval:
- The risks to subjects are minimized as much as possible.
- The risks to subjects are reasonable in relation to anticipated benefits.
- Selection of subjects is equitable.
- Informed consent will be sought from each prospective subject or their legally authorized representative and appropriately documented.
- The research plan makes provisions for monitoring the data collected to ensure safety of the subjects.
- Provisions are in place to protect the privacy of subjects and maintain confidentiality of data.
- When some or all of the subjects are likely to be vulnerable to coercion or undue influence, such as children, prisoners, individuals with impaired decision-making capacity, or economically or educationally disadvantaged persons, additional safeguards have been included in the study to protect the rights and welfare of these subjects.
These criteria ensure that the privacy and welfare of the research participants are adequately protected. All faculty and students engaged in such research should submit requests for IRB approval prior to beginning their work. This applies to all research activities, even those already approved by another institution.
For a complete description of all of the topics the IRB takes into consideration when reviewing a research activity, go to the Reviewing Research page.