Projects
Research in the Thomas Lab is centered around immunology, infectious diseases, molecular and computational biology.
Current research focus is on:
- systems immunology and molecular evolution of plasmodium, schistosomes and coronaviruses;
- genomic deconvolution of disease adaptation footprints in bovine trypanosomosis;
- transcriptomic dissection of epigenetically-reprogrammed parasites in leishmaniasis and metagenomics of cutaneous leishmaniasis;
- genetic architecture and population structure of sickle cell disease.
We utilize wet lab and computational biology tools to elucidate disease challenges related to susceptibility, tolerance, immunity and severity in each of these systems and population groups. Our activities are premised on training, mentoring and capacity development, including presentations at the Annual Biomedical Research Conference for Minority Students (ABRCMS), American Association of Immunologists conference and other professional society meetings.
A variety of techniques and approaches are used in the lab encompassing multiple disciplines including functional biology, gene expression, molecular biology, immunology, parasitology, computational biology and population genetics, amongst others.
Students interested in research should contact Dr. Thomas.
Systems Immunology and Molecular Evolution of Plasmodium, Schistosomes and Coronaviruses
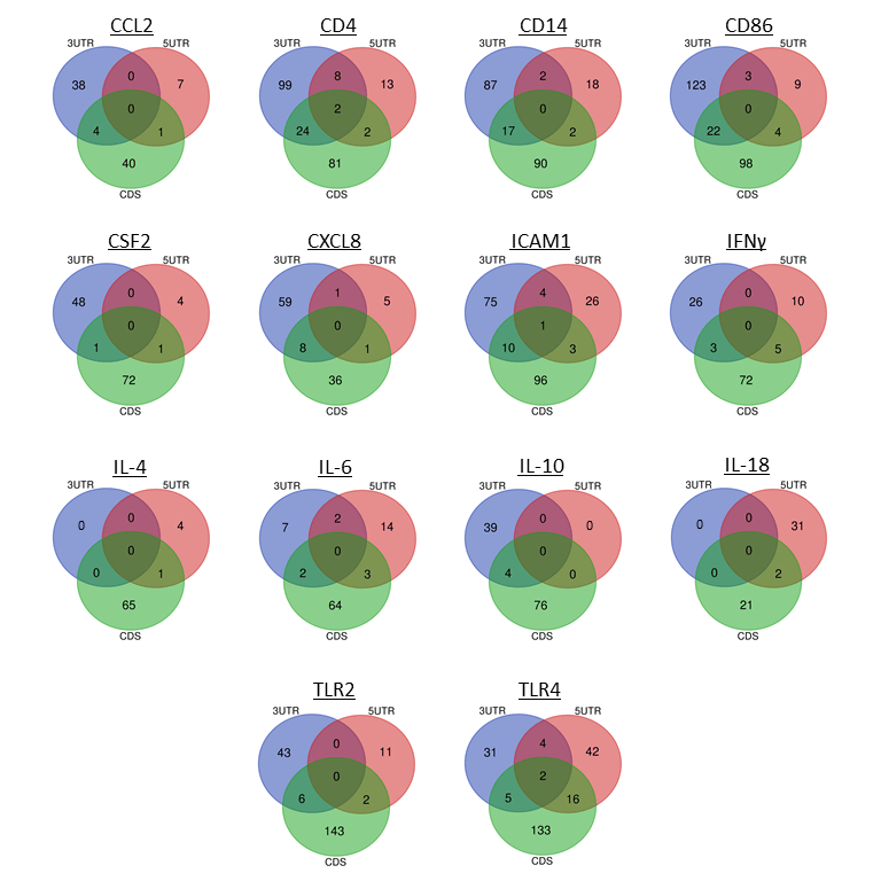
Venn Diagrams for target bovine mastitis genes showing the number of miRNA binding to the 3’ UTR, CDS, and 5’ UTR. The middle region of each diagram represents miRNA that bind to all three regions of the target gene (From Tucker et al., 2021; 10.1038/s41598-021-01280-9)
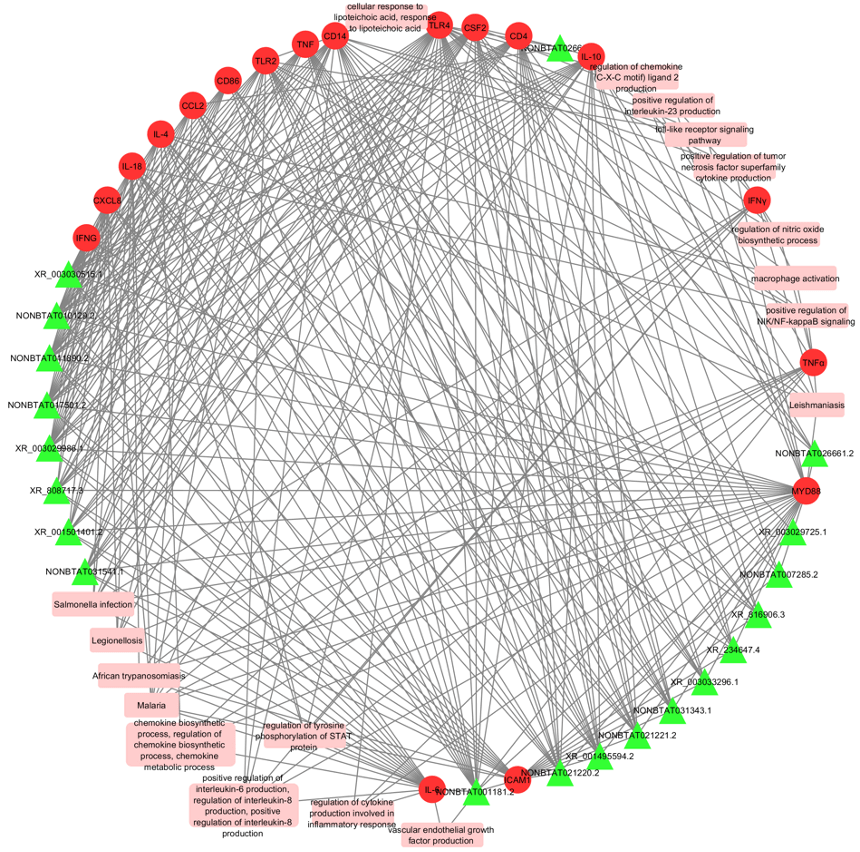
A network incorporating lncRNA, their target genes and corresponding gene ontologies. The green triangles are the lncRNA, red circles represent the target bovine mastitis genes, and the pink rectangles are the biological processes, molecular functions, cellular components, and pathways (Cytoscape version 3.7.2; https:// cytoscape.org/). (From Tucker et al., 2021; 10.1038/s41598-021-01280-9)

Mean parasitemia among malaria patients with CD209 (snp: ‐383A > G; rs4804803) wild type, heterozygous and mutant variants. Odds ratios were calculated by Fisher’s two‐tailed exact tests. A p‐value < 0.05 was considered significant. (From Morenikeji et al., 2020; 10.3390/microorganisms8020158)
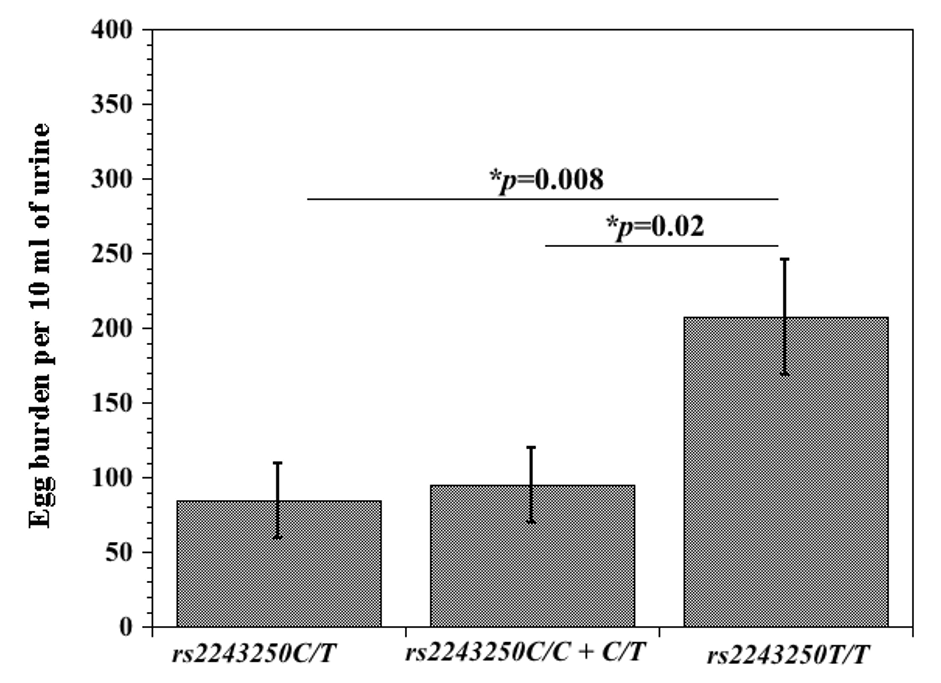
Mean egg burden between IL-4 (rs2243250) variants among schistosome-infected children. Urine samples were transported to the laboratory for microscopic examination of characteristic S. haematobium eggs, using the nucleopore filtration technique. Ten mls of well-mixed urine was aspirated and carefully forced through a filter membrane; filter was removed and placed on a slide, covered with cover slip and examined under a light microscope. The number of eggs on the entire filter was counted and recorded as the number of eggs per 10ml urine. rs2243250 C/T or rs2243250 C/C+C/T variants versus rs2243250 T/T) variant, showing significant difference in egg burden (p=0.008 and p=0.02 respectively) (From Adedokun et al., 2018; 10.1016/j.meegid.2018.07.012)
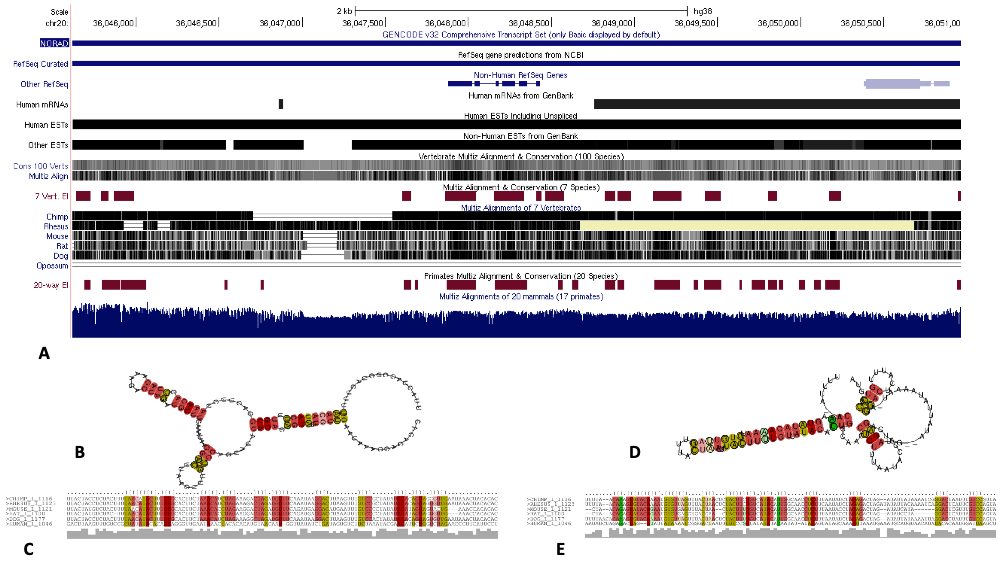
Thermodynamic stability and evolutionary conservation of lncRNA NORAD. To gain insight into the thermodynamic stability and evolutionarily conserved secondary structure of lncRNA NORAD, we carried out a multiple sequence alignment of six mammals including, input into RNAzWebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAz/RNAz.cgi) using default parameters. Server uses dynamic programming algorithm to determine encoding base-pair probabilities and RNA folding. A predicted RNA structure from the MSA with probability higher than 0.5 (P>0.5) is considered strong evidence for a structural RNA and evolutionary conservation. A, Annotated reference sequence of NORAD its conservation across several mammalian genome. B, Minimum free energy (MFE) structure encoding base-pair probabilities. C, Centroid structure drawing encoding base-pair probabilities. D, Mountain plot representation of the MFE structure, the thermodynamic ensemble of RNA structures, the centroid structure and the positional entropy for each position (From Morenikeji et al., 2021; 10.3389/fbioe.2020.582953)
Genomic Deconvolution of Disease Adaptation Footprints in Bovine Trypanosomosis

Multiple sequence alignment of CD14 promoter regions between 14 mammalian species; alignment is conserved within two groups separated into ruminants and non-ruminants. The CD14 protein sequence demonstrates significant variability of both percentage identity and similarity across the 14 species, despite common evolutionary origin (From Morenikeji and Thomas, 2019; 10.7717/peerj.7325)
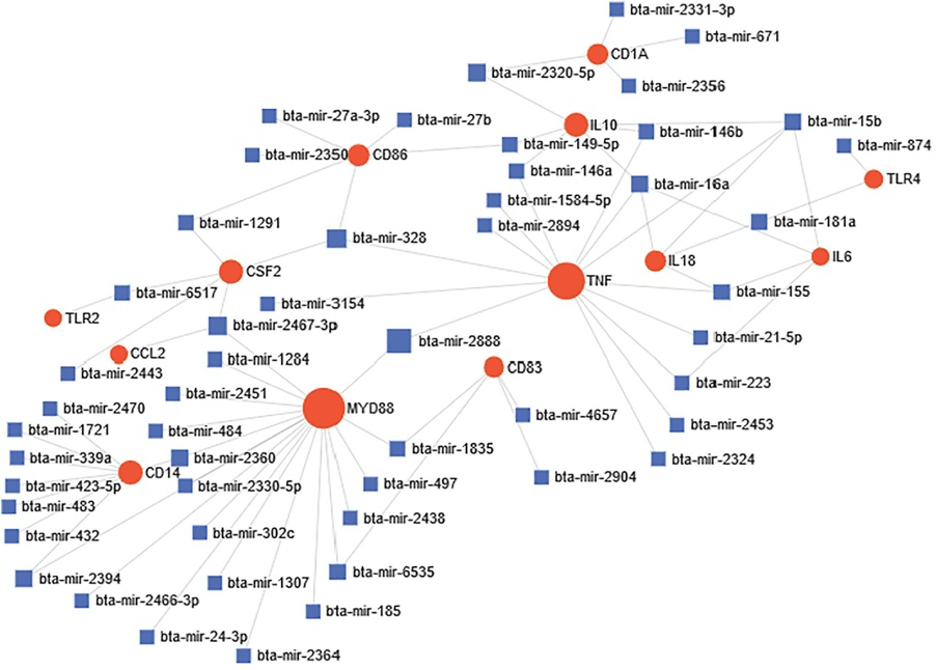
Predicted miRNA-target gene network during bovine trypanosomosis, showing significant connection between 13 genes and 54 miRNAs. Circular node shape (red color) represents significantly connected genes during disease. Node size simulates the number of interactions. miRNAs are represented by square shape and color blue. Most important miRNA nodes joined one or two genes together, thereby affecting their expression simultaneously (From Morenikeji et al., 2019; 10.3389/fmicb.2019.02010)
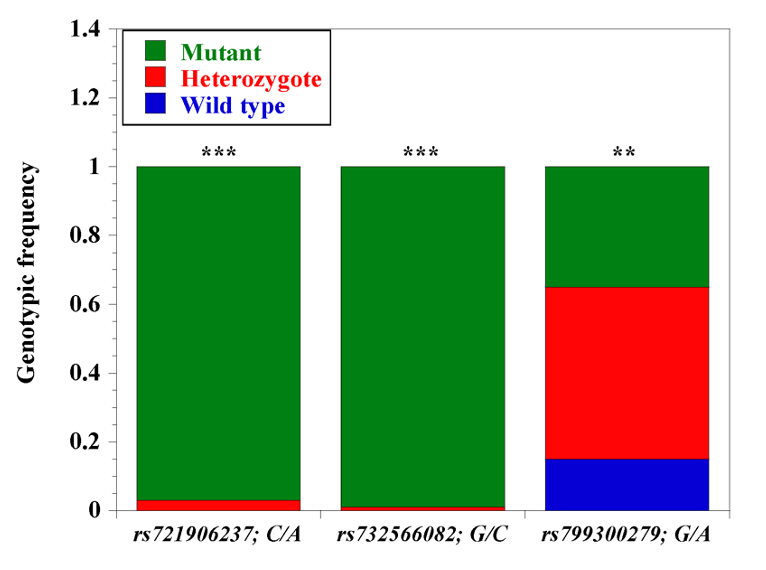
Genotypic frequency of three selected N’Dama-specific CD14 gene promoter single nucleotide polymorphisms. We elucidated the genetic diversity of 3 (out of 13) selected N’Dama-specific SNPs (n=103) using Taqman SNP genotyping assay. Blue bars: wild type variant; red bar: heterozygotes; green bars: mutant variants. The three SNPs are rs721906237; C/A; rs732566082, G/C; and rs799300279, G/A; *** significant (p<0.0001); * (p<0.01) (From Morenikeji et al., 2020; 10.3390/genes11010112).
Transcriptomic Dissection of Epigenetically-reprogrammed Parasites in Leishmaniasis and Metagenomics of Cutaneous Leishmaniasis
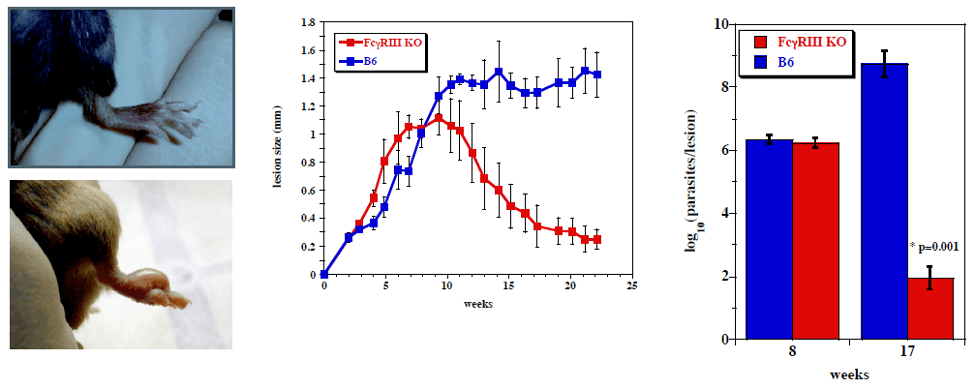
Uninfected and Leishmania-infected footpads (5x106 stationary-phase L. mexicana promastigotes). Differences in lesion size (p<0.05) and parasite burdens (p<0.01) between FcγRIII KO mice (which control parasite numbers and resolve L. mexicana lesions and C57BL/6 wild type, which produces chronic disease is shown at times indicated (From Thomas and Buxbaum, 2008)
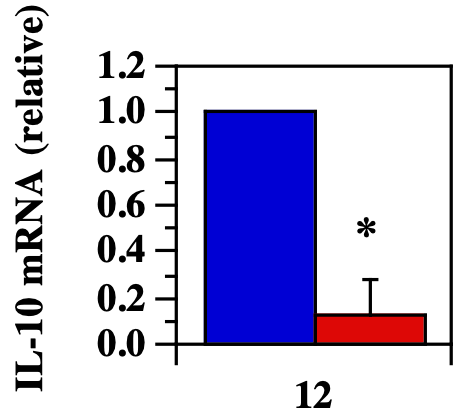
FcγRIII KO L. mexicana infected mice showed diminished lesion IL-10 production versus B6 wild-type mice. RNA purification from FcγRIII KO and B6 WT L. mexicana infected footpads and IL-10 quantification from both infection groups. (From Thomas and Buxbaum, 2008)

Anti-leishmanial IgG1 and IgG2a from serum of L. mexicana-infected mice bound to the surface of axenic amastigotes were used for bone marrow-derived infections. Such macrophages, prepared from B6 and FcγRIII KO mice were incubated under varying conditions (as shown) for 20 hrs. Macrophages were incubated with or without anti-IL-10R to block IL-10 uptake and signaling. We show a robust IL-10 induction (left) and IL-12p40 suppression (right), significantly boosted by the addition of both LPS and anti-IL-10R to infect B6 and FcγRIII KO bone marrow derived macrophages (Thomas and Buxbaum, 2008).
Genetic Architecture and Population Structure of Sickle Cell Disease

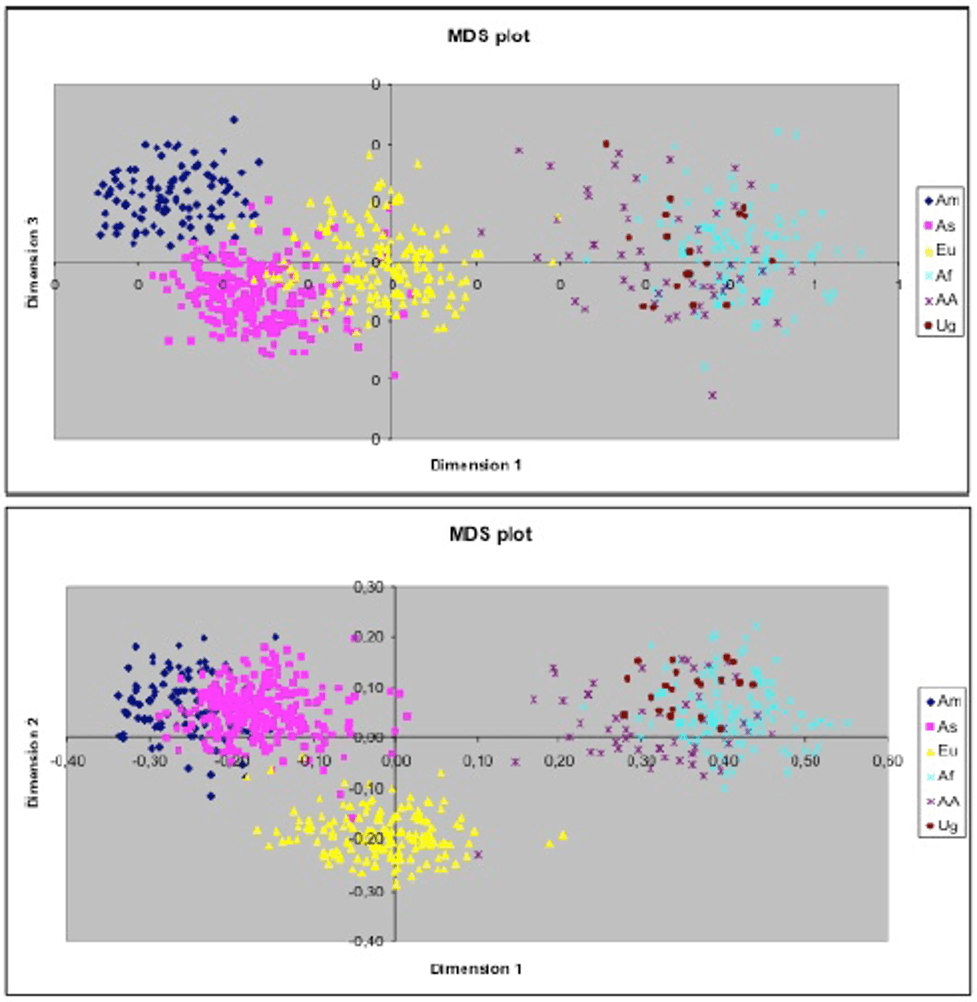
Two-dimensional plots of the first, second and third dimension obtained from an MDS analysis performed with an IBS distance matrix computed between pairs of individuals. Centroids of the four continental parental populations, using the HGDP-CEPH database, are marked by crosses (Am: Caucasian Americans, As: Asians; Eu: Europeans; Af: Africans; AA: African Americans; Ug: Ugandans)
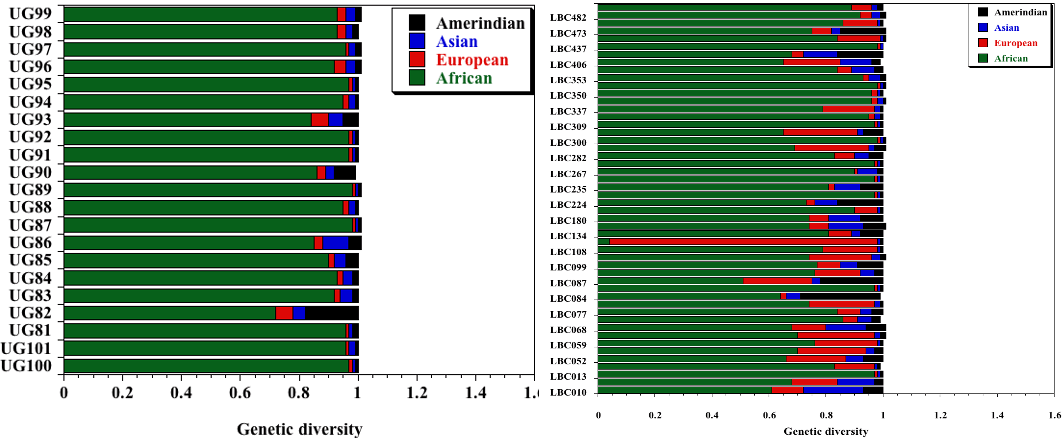
Individual continental contributions to selected African (left) and African American (right) gene pool (unpublished data); reveals significant admixture in the latter but not the former. The degree of genetic admixture in the African American sickle cell patients will support deconvoluting the population structure of the disease compared to that among African sickle cell disease group.