Research Areas
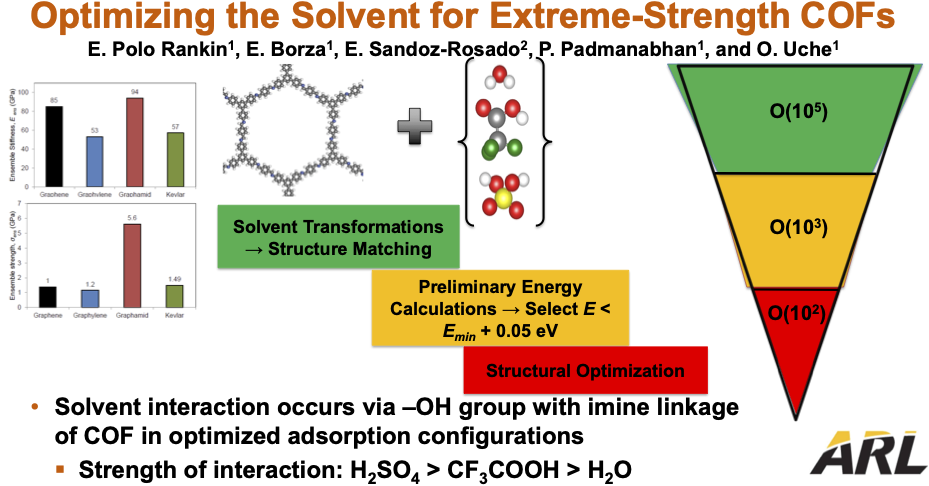
Two-dimensional polymers (or COFs) have shown promise as candidates for next-generation extreme strength materials as they combine the advantages of traditional Kevlar and more recent graphene-like materials. In order to manufacture these materials using high-throughput solution-type processing, one must dissolve large quantities of material in a suitable solvent. In this study, we utilize quantum mechanical calculations at small length scales as well as molecular dynamics simulations at intermediate length scales to understand the nature of interactions between the 2-D polymer and different solvents. Adsorption configurations, binding energies, and charge transfer between the polymer and solvent molecules will be used to evaluate the strength of interactions and eventually, to predict optimal solvents for this material.
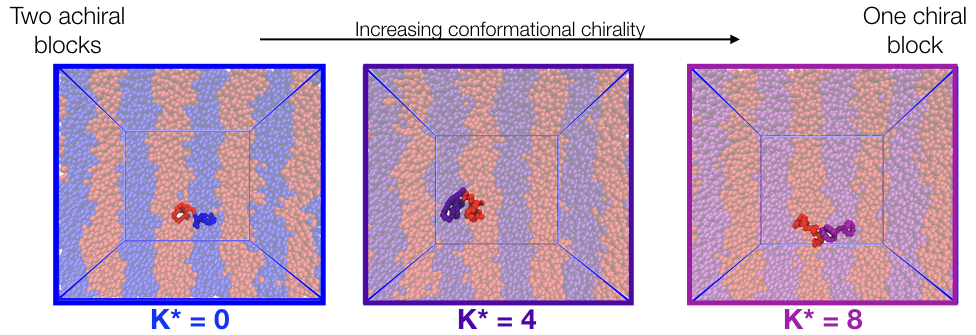
Chirality is the feature that an object cannot be superimposed on its mirror image by rotations or translations. In block copolymers, hierarchical chirality is possible at the monomer scale, the conformational scale, and the mesoscale. But the mechanisms o chirality transfer from the monomer all the way up to the mesoscale is not yet fully understood. Using simulations, we investigate the thermodynamics of chiral block copolymers, starting with the simplest lamellar morphology, aiming to understand mechanisms of chirality (or lack thereof) during self-assembly. Our preliminary results suggest that chains sacrifice conformational chirality to favor the formation of mesoscale structure.

Molecular-based design of materials rests on the ability to validate molecular models with experimental electrochemical measurements at the macroscopic level. In that regards, we work on building a bridge to link observed macroscopic behavior to the physics at the molecular scale during electrochemical processes. As a first case of application, we focus our efforts on electrical double layer (EDL) formation within nanostructures. We develop algorithms and modeling schemes in order to describe and predict formation of electrical double layer (EDL) within porous materials, and translate EDL structure into electrochemical response. Simultaneously, we fabricate porous materials with tailored properties such as architecture and dimensions in order to perform macroscopic electrochemical measurements indicative of EDL behavior. We are increasingly closing the gap in scales and working towards direct validation of modeling predictions with experimental results. The final goal is to propose a framework and methodology for the molecular-based design of hierarchical materials and processes for advanced ion separations and electrochemical charge storage.
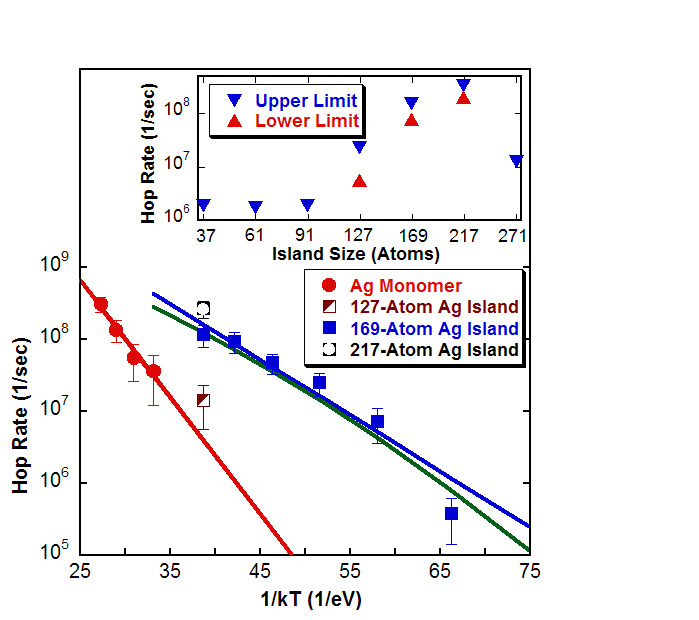
Investigations of the dynamics and behavior of materials at interfaces have major implications for epitaxial film growth and other similar processes. Our work in the area of interfacial phenomena has focused on studying the evolution of hetero-epitaxial metallic systems using parallel replica dynamics (PRD) and nudged elastic band methods and thus, elucidating diffusion mechanisms and energetics of such systems. Using the PRD technique enables the extension of molecular dynamics (MD) simulations to longer times and lower temperatures; thus, revealing lowest-barrier pathways for diffusion. Past results have indicated that certain two-dimensional islands of silver undergo extremely rapid one-dimensional diffusion on Cu(001) surfaces. The novel collective diffusion mechanism discovered herein involved vacancy diffusion on island edges coupled with simultaneous glide of the island core. It should be noted that hopping rates for these ‘‘magic-size’’ islands were shown to be orders of magnitude faster than hopping rates for single silver adatoms below 300K. These results suggest a possible modification in the picture of film growth of Ag on Cu(001) surfaces which have generally allowed for only single atom hops and thus neglected such complex cooperative mechanisms.

Naturally-occurring gas hydrates (in permafrost and marine sediments) have the potential to contribute as a carbon-based source to the increasing energy demand or as a source of methane (a potent greenhouse gas) if destabilized. Precise estimates of gas-hydrate global inventory and evaluation of their environmental impact requires models that accurately represent the physics of gas hydrates in marine sediments in particular. The goal of the project is to develop and validate predictive models for stability, formation and dissociation of gas hydrates in porous matrices. The hypothesis for the work is that thermodynamic stability and growth/dissociation of hydrates in sediments depends on the nature of the surface interactions and confinement effects. The final goal is to have models of increased accuracy built in layers that can be combined with reservoir simulators to accurately describe gas hydrate reservoirs and predict their environmental impact.
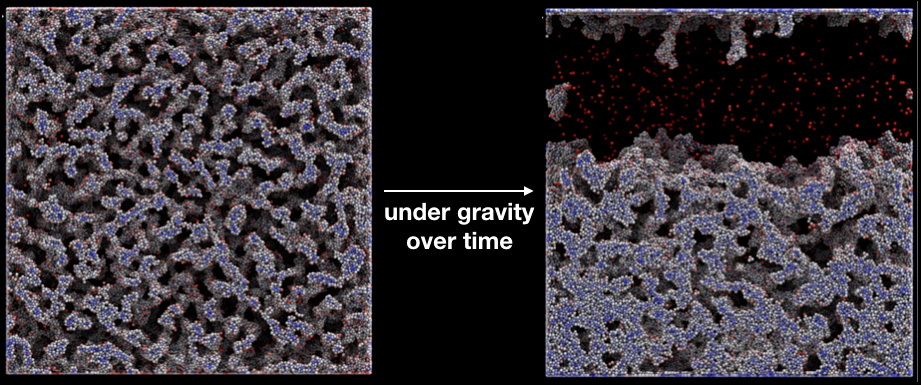
Disordered bicontinuous networks are formed via the aggregation of colloidal particles over a range of temperatures and volume fraction of the particles. These gels (networks) are stable mechanically but can also flow under applied forces, and reconstitute their network structure after cessation of flow. Thus, the colloidal gels are workhorse models for self-healing materials. In particular, their mechanical response under gravity has important implications in the shelf-life of these materials. The structure would seemingly be stable for some time, but would subsequently undergo dramatic collapse. We utilized large- scale simulation to model their assembly and flow, we studied the micromechanical origins of gel collapse and connected the macroscopic response of the gel to its bond evolution and phase behavior.

Precious metals and rare elements display high catalytic activities that have yet to be matched by upcoming catalyst formulations in most catalytic processes, for example water splitting. The goal of the research area is to identify, manipulate and inform on the links between catalyst architecture, molecular phenomena and composition at the nanoscale and catalytic activity. Moving away from improving catalytic activity via increasingly-complex chemical functionality requires knowledge of what structural properties of a catalyst may trigger key steps in a given catalytic reaction. A combination of experiments and modeling is used in order to identify the specific characteristics that can impart activity for hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) onto hierarchically-structured titanium/titanium dioxide (Ti/TiO2) electro-catalysts, the first model system used in the project. The hypothesis driving the work is that electro-catalytic activity can be imparted to Ti/TiO2 materials via control of the hierarchical structure of metal-core/semiconductor-coating via enabling the formation of critical intermediates and the onset of key reaction-mechanism steps. The final goal of this research area is the design framework and directives towards the tuning of architecture at all scales of a new class of catalysts.

Understanding catalyst structure/function relationships provides opportunities both to enhance current catalyst performance and to develop new catalysts. Recent advances in computational techniques make it possible to investigate such problems with similar accuracy but lower costs in comparison to experiments. Dr. Uche also applies ab-initio techniques to investigate problems in catalysis such as methane activation on various chalcogenide surfaces as well as in exchanged zeolite frameworks. Specifically, she and her collaborators have focused on the development of alternative “softer” catalysts for methane-to-olefin (MTO) production. First-principles density functional theory (DFT) calculations have been performed to help understand the reaction pathways and mechanisms, the nature of the active catalytic sites and the effects of the sulfide catalyst, as well as the reaction conditions on methane conversion and the selective formation of ethylene. Past results have demonstrated that lower (more controlled) conversions of methane as well as higher selectivities of methane to ethylene conversion were observed on sulfides with the strongest surface metal-sulfur bonds.

Ordered bicontinuous networks are made of one or more networks within a continuous matrix, and can be formed via the self-assembly of amphiphilic molecules. These structures have the advantage of being mechanically stable, highly porous, and possess isotropic (directionally-independent) properties. We target a special subset of ordered networks that have superior optical properties and can bend light in non-traditional ways for use in solar cells. In addition, these structures can serve as templates for potentially transformative materials with novel properties such as negative refractive index materials for invisibility cloaks. But such networks are difficult to synthesize in experiment due to their narrow range of stability in the phase diagram. We work on developing theoretical models to study these phase diagrams and devise strategies to design the macromolecular building blocks that result in the targeted structure.